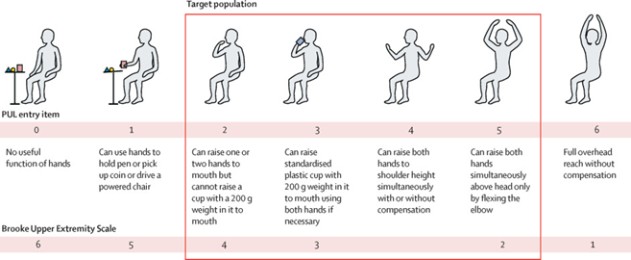
Performance of the Upper Limb (PUL entry items) (1)
(CAP-1002 current DMD target population)
| (1) | Image from HOPE-2 Lancet Publication (March 2022) |
Enrollment is underway for Cohort B, which is designed to enroll approximately 44 participants randomized to either CAP-1002 or placebo in a 1:1 ratio. A primary efficacy and safety analysis will be performed for each individual cohort at month 12, following 4 administrations of CAP-1002 or placebo. We plan to complete enrollment for Cohort B in the second quarter of 2024. Cohort B uses product manufactured at our San Diego facility.
The primary outcome measure of the HOPE-3 study will be the Performance of the Upper Limb (“PUL”) v2.0, a validated tool specifically designed for assessing high (shoulder), mid (elbow) and distal (wrist and hand) functions, with a conceptual framework reflecting weakness progression in upper limb function. HOPE-3 will also measure various secondary endpoints including cardiac function assessments.
Under our RMAT designation, in the third quarter of 2023, we met with the FDA in a Type-B meeting where we discussed our manufacturing plans in anticipation of potentially submitting a BLA application. In this meeting, we affirmed alignment with respect to our Phase 3, HOPE-3 program. Additionally, we discussed our plans with respect to commercial manufacturing activities, including our potency assay and other product release criteria to support commercialization. We plan to meet with FDA in the first quarter of 2024 to continue discussing our pathway to BLA. In the upcoming Type-B meeting, we intend to discuss our further CMC plans for commercial launch, if approved, with the aim of expediting our BLA submission pathway. Our ultimate goal is to file a BLA allowing for the use of CAP-1002 commercial product manufactured at our San Diego facility.
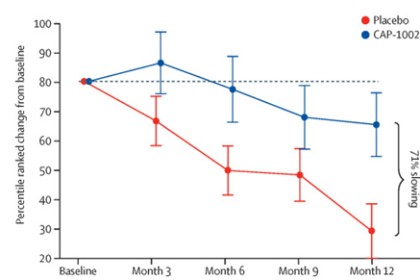
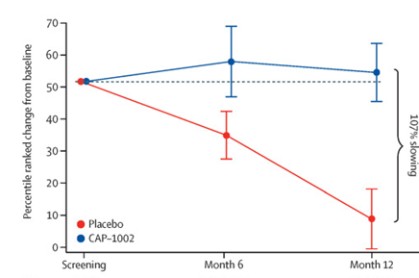
Phase 2 HOPE-2 Clinical Trial: HOPE-2 was a randomized, double-blind, placebo-controlled clinical trial conducted at multiple sites in the United States and was completed in 2021. The clinical trial was designed to evaluate the safety and efficacy of repeated, intravenous doses of CAP-1002, in boys and young men with evidence of skeletal muscle impairment regardless of ambulatory status. Approximately 90% of the patients in the study were non-ambulant and all patients were on a stable regimen of steroids. Demographic and baseline characteristics were similar between the two treatment groups. The final one-year results from HOPE-2 were published in The Lancet in March 2022, showing that the trial met its primary efficacy endpoint of the mid-level dimension of the PUL v1.2 (p=0.01) and additional positive endpoints of full PUL v2.0 (p=0.04). Although the PUL v1.2 for the mid-level was the primary endpoint established for the trial, we also conducted an analysis using the PUL v2.0 as the FDA suggested the use of the updated PUL v2.0 as the primary efficacy endpoint in support of a BLA. Left ventricular ejection fraction (LVEF), a global measure of cardiac pump function, decreased in the placebo group over time, but improved in the CAP-1002 group, showing a 107% slowing of the progression of cardiac disease (p=0.002). Additionally, the data suggested global improvements in cardiac function as measured by indexed volumes (LVESV, LVEDV). These are surrogate measures of cardiac function and are considered significant in relevance to long-term outcomes. Furthermore, the data showed a reduction in the biomarker CK-MB, an enzyme that is only released when there is cardiac muscle cell damage. In normal human subjects, there is typically no CK-MB measurable in the blood. It is well accepted that continuous muscle cell damage in DMD leads to pathologically high enzyme levels associated with cardiac muscle cell loss. To our knowledge, this is the first clinical study in DMD that correlates cardiac functional stabilization with a reduction of a biomarker of cell damage. With the exception of steroids, preservation of function in DMD is uncommon. The results of the placebo patients were consistent with natural history, but in the treated group, most patients were stable or improved on these endpoints throughout the one-year treatment period. CAP-1002 was generally safe and well tolerated throughout the study. With the exception of hypersensitivity
7